Spatial perturbation response visualization (replot.py) - ganitumab sensitivity, 50um-Local Model
import sys
import re
import pickle
import numpy as np
from scipy.stats import rankdata
from datasets import load_from_disk
from sklearn.cluster import KMeans
import matplotlib.pyplot as plt
import scanpy as sc
import pandas as pd
import math
import os
import gzip
import bz2
import lzma
if __name__=="__main__":
pkl_file = sys.argv[1]
with open(pkl_file, "rb") as f:
plot_data = pickle.load(f)
coord_x = plot_data["coord_x"]
coord_y = plot_data["coord_y"]
patients = plot_data["patient"]
leiden = plot_data["leiden"]
umap = plot_data["umap"]
rbp_score = plot_data["rbp_score"]
gene_names = plot_data["gene_names"]
cluster_tables = plot_data["cluster_tables"]
missing_by_sample = plot_data["missing_by_sample"]
adata = plot_data["adata"]
ig, axes = plt.subplots(1, 2, figsize=(12, 5))
sc.pl.umap(adata, color='leiden', ax=axes[0], show=False, size=20, legend_loc='on data')
axes[0].set_title("UMAP — Leiden Clusters")
sc.pl.umap(adata, color='patient', ax=axes[1], show=False, size=20)
axes[1].set_title("UMAP — Patient Labels")
plt.tight_layout()
plt.savefig("top100_umap_leiden_patient.pdf", dpi=300)
plt.close()
ig, axes = plt.subplots(1, 2, figsize=(12, 5))
sc.pl.umap(adata, color='leiden', ax=axes[0], show=False, size=20, legend_loc='on data')
axes[0].set_title("UMAP — Leiden Clusters")
sc.pl.umap(adata, color='patient', ax=axes[1], show=False, size=20)
axes[1].set_title("UMAP — Patient Labels")
plt.tight_layout()
plt.savefig("top100_umap_leiden_patient.pdf", dpi=300)
plt.close()
umap = adata.obsm["X_umap"]
x = umap[:, 0]
y = umap[:, 1]
unique_patients = np.unique(patients)
palette = sc.pl.palettes.default_20
colors = {p: palette[i % len(palette)] for i, p in enumerate(unique_patients)}
n = len(unique_patients)
cols = 6 # change to 4, 5, etc. if you prefer
rows = int(np.ceil(n / cols))
fig, axes = plt.subplots(rows, cols, figsize=(5*cols, 5*rows))
axes = np.array(axes).reshape(rows, cols)
for ax, patient in zip(axes.flatten(), unique_patients):
mask = (patients == patient)
ax.scatter(x[~mask], y[~mask], s=6, c="#dddddd", alpha=0.3, edgecolors="none")
ax.scatter(x[mask], y[mask], s=12, c=colors[patient], label=patient, edgecolors="none")
ax.set_title(f"Patient: {patient}", fontsize=12)
ax.set_xticks([])
ax.set_yticks([])
ax.axis("off")
for ax in axes.flatten()[n:]:
ax.axis("off")
plt.tight_layout()
plt.savefig("top100_umap_by_patient_panels.png", dpi=300)
plt.close()
cluster_labels = leiden
unique_clusters = sorted(np.unique(cluster_labels))
print(unique_clusters)
n_clusters = len(unique_clusters)
cols = 3
rows = math.ceil(n_clusters / cols)
fig, axes = plt.subplots(rows, cols, figsize=(4 * cols, 3 * rows))
axes = np.array(axes).reshape(rows, cols)
for ax in axes.flatten()[n_clusters:]:
ax.axis("off")
for ax, cl in zip(axes.flatten(), unique_clusters):
df = cluster_tables[cl]
ax.axis("off")
ax.set_title(f"Cluster {cl}", fontsize=10)
cell_text = [[row["gene"], f"{int(row['count'])}", f"{row['fraction_cells']:.2f}"] for _, row in df.iterrows()]
col_labels = ["gene", "count", "fraction_cells"]
table = ax.table(cellText=cell_text, colLabels=col_labels, loc="center")
table.auto_set_font_size(False)
table.set_fontsize(8)
table.scale(1.0, 1.2) # tweak if needed
plt.tight_layout()
plt.savefig("top100_top_genes_per_cluster_tables.pdf", dpi=300)
plt.close()
tables = cluster_tables # or top100_cluster_tables
unique_clusters = sorted(tables.keys(), key=lambda x: int(x))
n_clusters = len(unique_clusters)
cols = 5
rows = int(math.ceil(n_clusters / float(cols)))
plt.figure(figsize=(1.5 * cols, 2.5 * rows))
fig, axes = plt.subplots(rows, cols, figsize=(1.5 * cols, 2.5 * rows), sharex=False, sharey=False)
axes = np.array(axes).reshape(rows, cols)
for ax, cl in zip(axes.flatten(), unique_clusters):
genes = tables[cl]["gene"]
fracs = tables[cl]["fraction_cells"]
y = np.arange(len(genes))
ax.barh(y, fracs)
ax.set_yticks(y)
ax.set_yticklabels(genes)
ax.invert_yaxis()
ax.set_title(f"Cluster {cl}", fontsize=10)
ax.set_xlabel("Fraction of cells")
ax.set_ylim(-0.5, len(genes) - 0.5)
for ax in axes.flatten()[n_clusters:]:
ax.axis("off")
plt.tight_layout()
plt.savefig("horizontal_barplots_by_cluster.png", dpi=300)
plt.close()
'''
vv_max = max([max(tables[cl]["fraction_cells"]) for cl in unique_clusters])
for ax, cl in zip(axes.flatten(), unique_clusters):
genes = np.array(tables[cl]["gene"])
fracs = np.array(tables[cl]["fraction_cells"], dtype=float)
counts = np.array(tables[cl]["count"], dtype=float)
order = np.argsort(fracs)[::-1]
genes = genes[order]
fracs = fracs[order]
counts = counts[order]
y = np.arange(len(genes))
sizes = counts/2
ax.scatter(np.zeros_like(y), y, s=sizes, c=fracs, cmap="inferno", vmin=0.0, vmax=vv_max)
ax.set_yticks(y)
ax.set_yticklabels(genes)
ax.set_xticks([])
ax.invert_yaxis()
ax.set_title(f"C{cl}", fontsize=10)
for ax in axes.flatten()[n_clusters:]:
ax.axis("off")
plt.tight_layout()
plt.savefig("bubbleplots_by_cluster.png", dpi=300)
plt.close()
'''
unique_patients = np.unique(patients)
unique_clusters = sorted(np.unique(cluster_labels))
palette = sc.pl.palettes.default_20
cluster_color_map = {cl: palette[i % len(palette)] for i, cl in enumerate(unique_clusters)}
n_patients = len(unique_patients)
cols = 5 # tweak grid
rows = int(math.ceil(n_patients / cols))
fig, axes = plt.subplots(rows, cols, figsize=(4 * cols, 4 * rows), sharex=False, sharey=False)
axes = np.array(axes).reshape(rows, cols)
for ax, patient in zip(axes.flatten(), unique_patients):
mask_patient = (patients == patient)
for cl in unique_clusters:
mask = mask_patient & (cluster_labels == cl)
if not np.any(mask):
continue
ax.scatter(coord_x[mask], coord_y[mask], s=20, c=cluster_color_map[cl], edgecolors="none")
ax.set_title(f"Patient: {patient}", fontsize=10)
ax.set_xticks([])
ax.set_yticks([])
ax.invert_yaxis()
for ax in axes.flatten()[n_patients:]:
ax.axis("off")
# Global legend for clusters
handles = [plt.Line2D([0], [0], marker="o", linestyle="", markersize=15, color=cluster_color_map[cl],
label=f"Cluster {cl}") for cl in unique_clusters]
fig.legend(handles=handles, loc="center right", title="Leiden clusters", bbox_to_anchor=(0.98, 0.5), frameon=True)
plt.tight_layout()
plt.subplots_adjust(right=0.85)
plt.savefig("top100_spatial_by_patient_clusters.png", dpi=300)
plt.close()
full_sample = plot_data["full_sample"]
full_x = plot_data["full_x"]
full_y = plot_data["full_y"]
full_barcode = plot_data["full_barcode"]
barcodes = adata.obs["barcode"]
sub_sample = patients
sub_barcode = barcodes
sub_x = coord_x
sub_y = coord_y
matched_samples = set(np.intersect1d(np.unique(full_sample), np.unique(sub_sample)))
full_by_sample = {}
for i in range(full_sample.shape[0]):
sid = full_sample[i]
if sid not in matched_samples:
continue
if sid not in full_by_sample:
full_by_sample[sid] = {"barcode": [], "x": [], "y": []}
full_by_sample[sid]["barcode"].append(full_barcode[i])
full_by_sample[sid]["x"].append(full_x[i])
full_by_sample[sid]["y"].append(full_y[i])
sub_barcode_set_by_sample = {}
for i in range(sub_sample.shape[0]):
sid = sub_sample[i]
if sid not in matched_samples:
continue
if sid not in sub_barcode_set_by_sample:
sub_barcode_set_by_sample[sid] = set()
sub_barcode_set_by_sample[sid].add(sub_barcode[i])
missing_by_sample = {}
for sid in matched_samples:
if sid not in full_by_sample:
continue
fb = full_by_sample[sid]
sub_set = sub_barcode_set_by_sample.get(sid, set())
miss_x = []
miss_y = []
for b, x, y in zip(fb["barcode"], fb["x"], fb["y"]):
if b not in sub_set:
miss_x.append(x)
miss_y.append(y)
if len(miss_x) > 0:
missing_by_sample[sid] = (np.array(miss_x), np.array(miss_y))
scores = rbp_score
unique_patients = sorted(np.unique(patients))
n_patients = len(unique_patients)
vmin = np.percentile(scores, 5)
vmax = np.percentile(scores, 90)
cols = 5 # tweak as needed
rows = int(np.ceil(n_patients / float(cols)))
fig, axes = plt.subplots(rows, cols, figsize=(4 * cols, 4 * rows), sharex=False, sharey=False)
axes = np.array(axes).reshape(rows, cols)
last_scatter = None
new_patients = ["1142243F", "1160920F", "CID4290", "CID4535", "GSM6433586", "GSM6433589", "GSM6433590",
"GSM6433591", "GSM6433592", "GSM6433599", "GSM6433600", "GSM6433601", "GSM6433602", "GSM6433613", "GSM6433618"]
new_patients = [nn for nn in new_patients if nn in unique_patients]
remain_patients = [un for un in unique_patients if un not in new_patients]
unique_patients = new_patients + remain_patients
for ax, patient in zip(axes.flatten(), unique_patients):
#ax.set_facecolor("black")
mask = (patients == patient)
if not np.any(mask):
ax.axis("off")
continue
sample_ids_p = set(np.unique(sub_sample[mask]))
for sid in sample_ids_p:
if sid in missing_by_sample:
mx, my = missing_by_sample[sid]
ax.scatter(mx, my, s=10, c="grey", edgecolors="none", alpha=0.6)
last_scatter = ax.scatter(coord_x[mask], coord_y[mask], c=scores[mask], s=20, cmap="inferno", vmin=vmin, vmax=vmax)
ax.set_title(f"Patient: {patient}", fontsize=10)
ax.invert_yaxis() # often needed for Visium-style coords
ax.set_xticks([])
ax.set_yticks([])
for ax in axes.flatten()[len(unique_patients):]:
ax.axis("off")
fig.subplots_adjust(right=0.88)
cax = fig.add_axes([0.90, 0.15, 0.02, 0.70])
if last_scatter is not None:
fig.colorbar(last_scatter, cax=cax, label="RBP score")
plt.tight_layout(rect = [0, 0, 0.88, 1])
plt.savefig("rbp_score_spatial_by_patient.png", dpi=300)
plt.close()
Running Command
python3 replot.py replot_bundle_250929062100_run-d565b1cd.pkl
This loads a pickle file containing the results.
Results
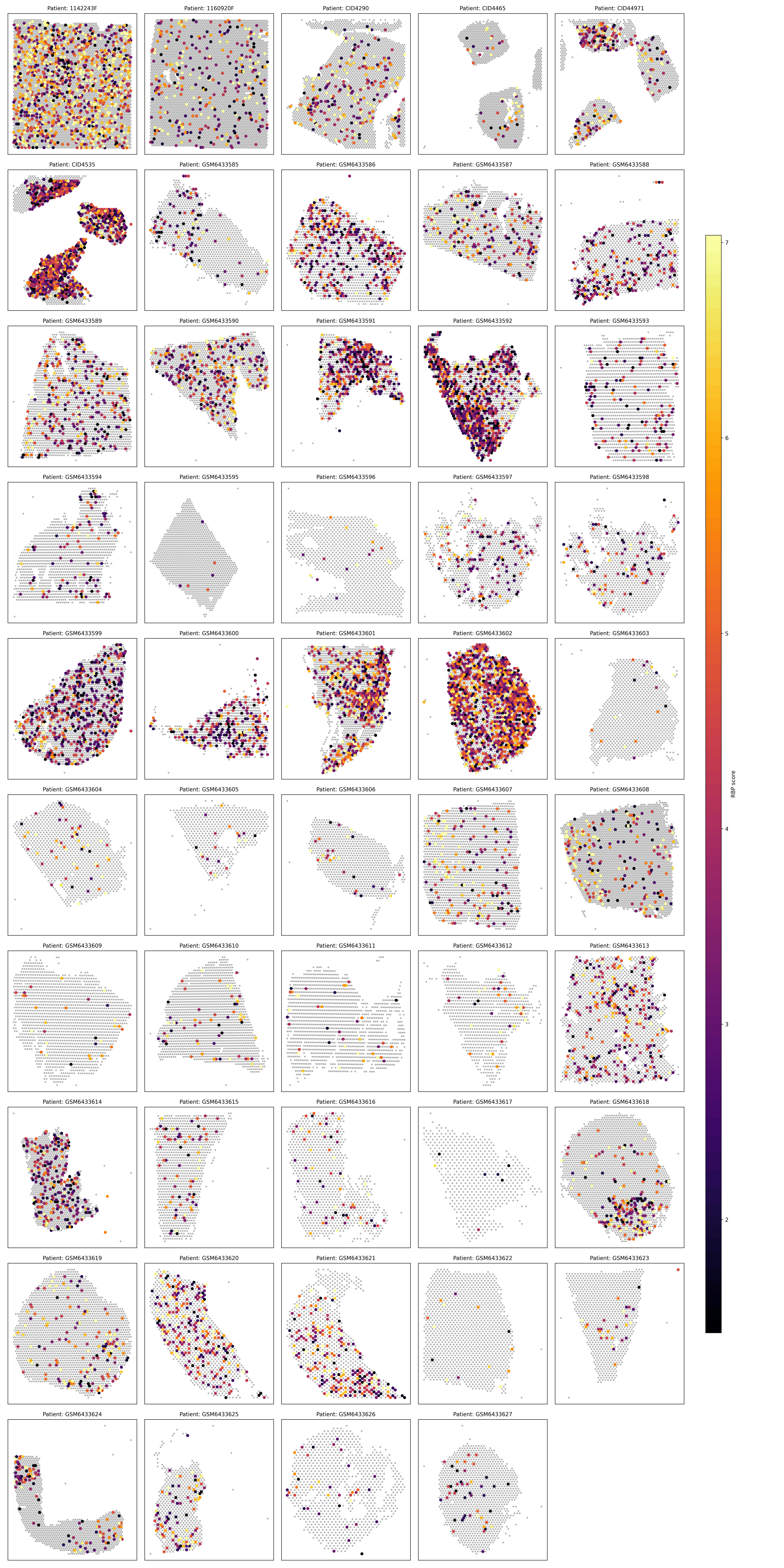
rbp_score_spatial_by_patient.png:
Shows the sensitivity score of ganitumab treatment per spot per spatial transcriptomic sample. Here they are TNBC samples. 3 samples specifically exhibit higher sensitivity score than rest.
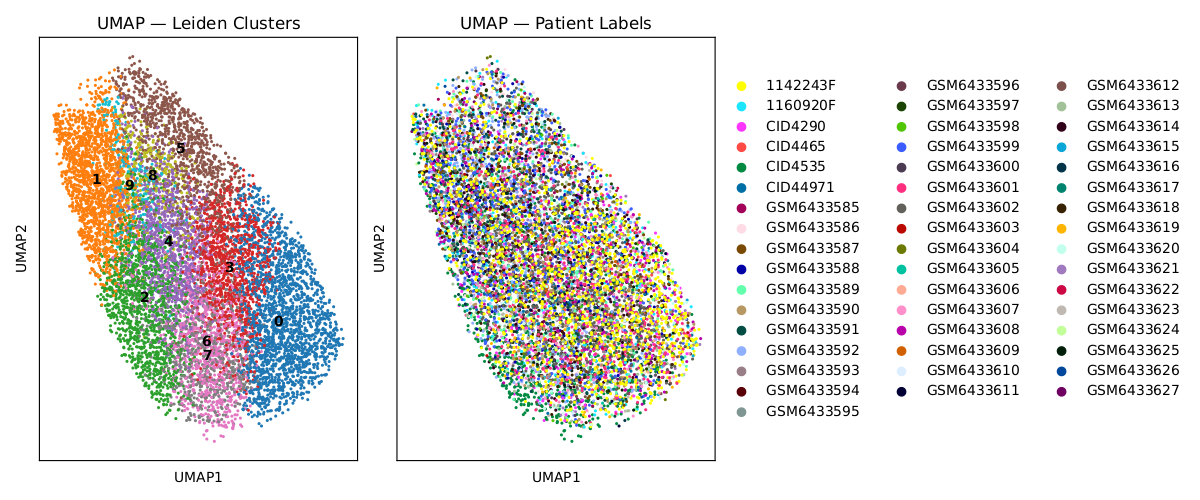
top100_umap_leiden_patient.png:
Shows how ST spots cluster by sensitivity profiles. Spot-by-gene response matrix (for top 100 response genes and IGF1R-expressing spots) was used for Leiden clustering followed by UMAP visualization.
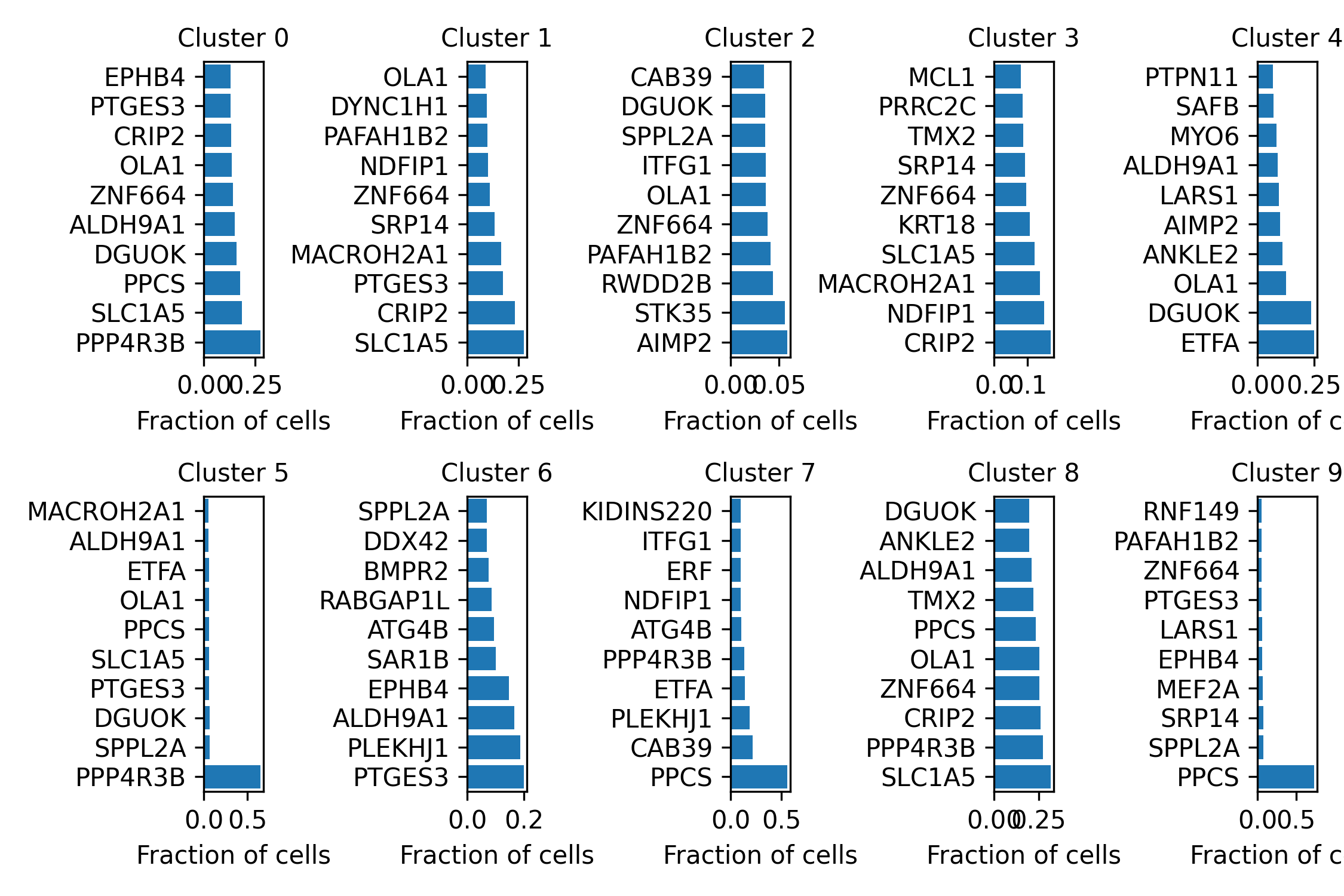
horizontal_barplots_by_cluster.png:
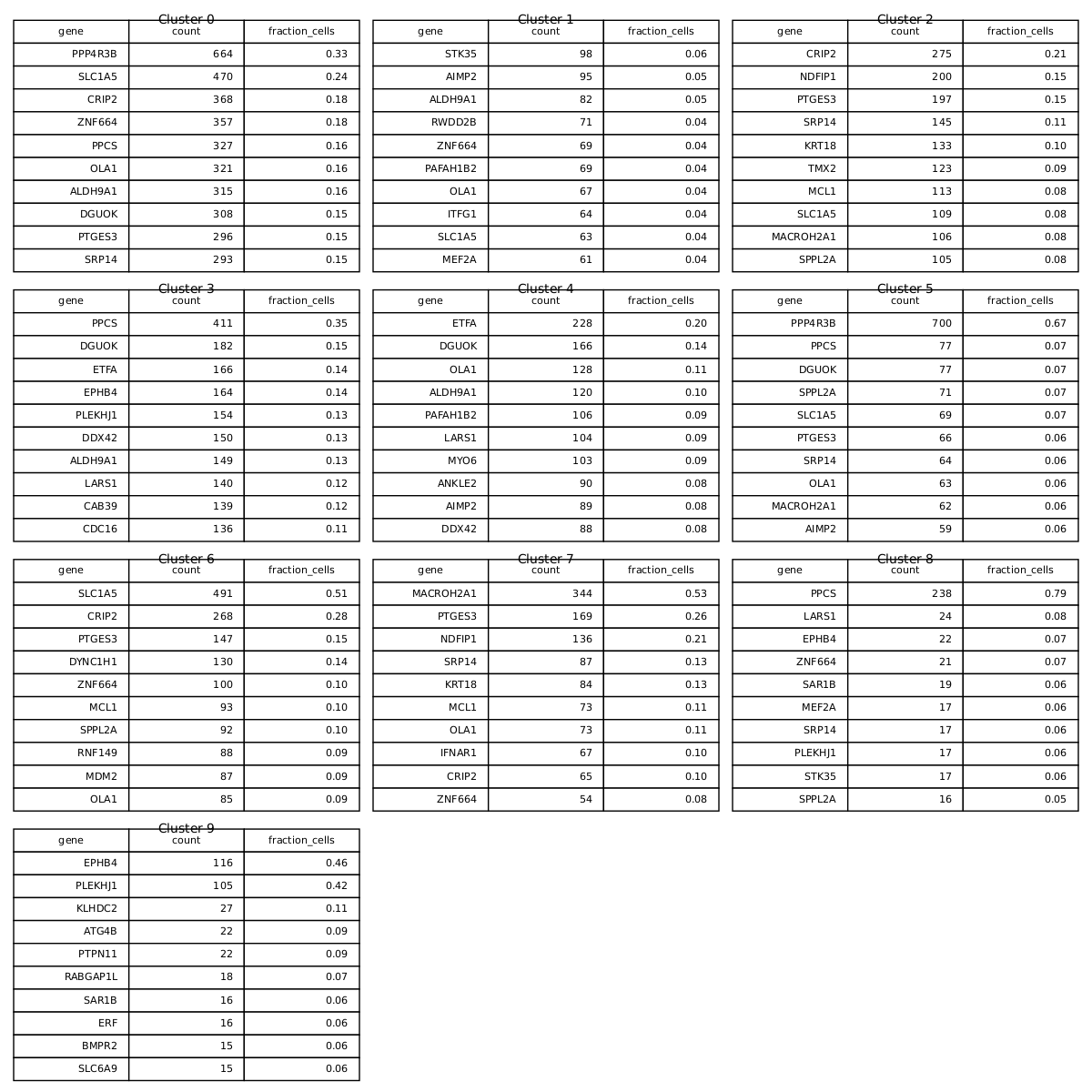
Shows the top 10 marker genes of each sensitivity spot-cluster. Marker genes represent sensitive genes.
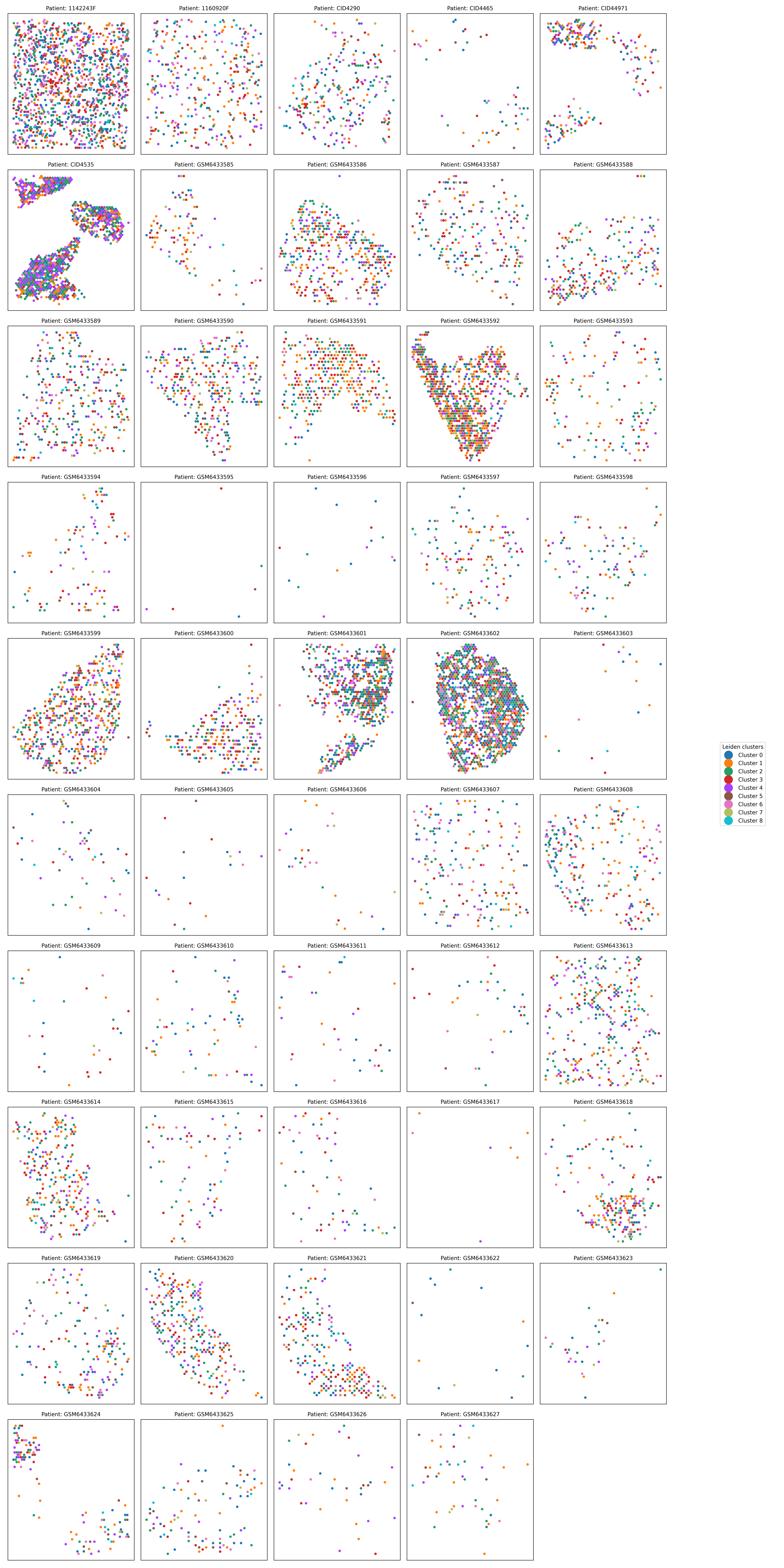
top100_spatial_by_patient_clusters.png:
Shows how sensitivity clusters distribute spatially across ST samples.
top100_top_genes_per_cluster_tables.png:
Shows the source data for the marker gene horizontal bar plot above.