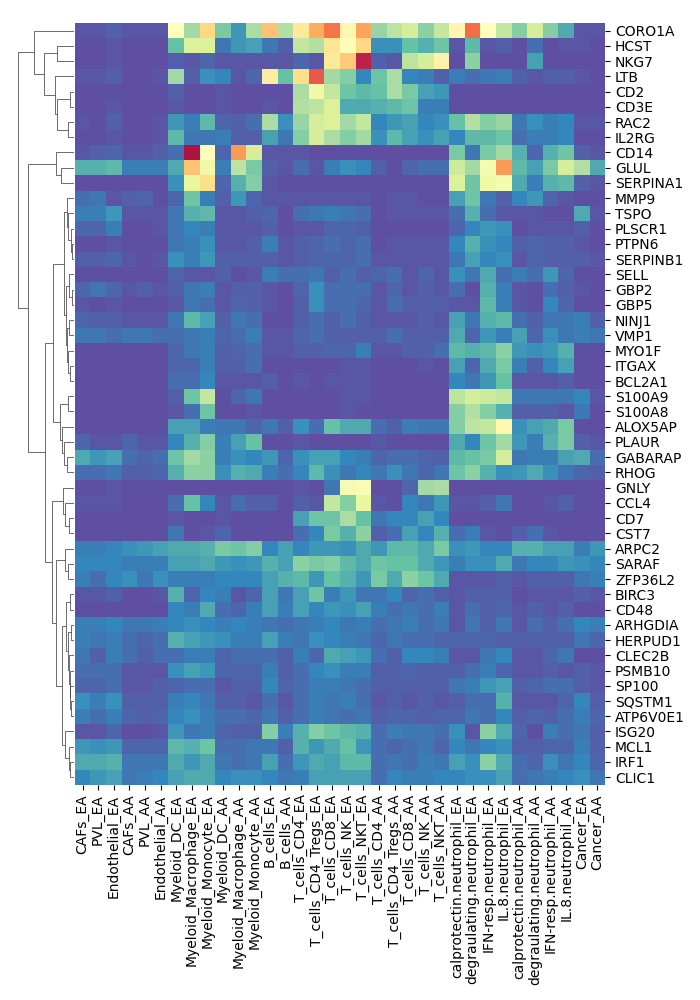
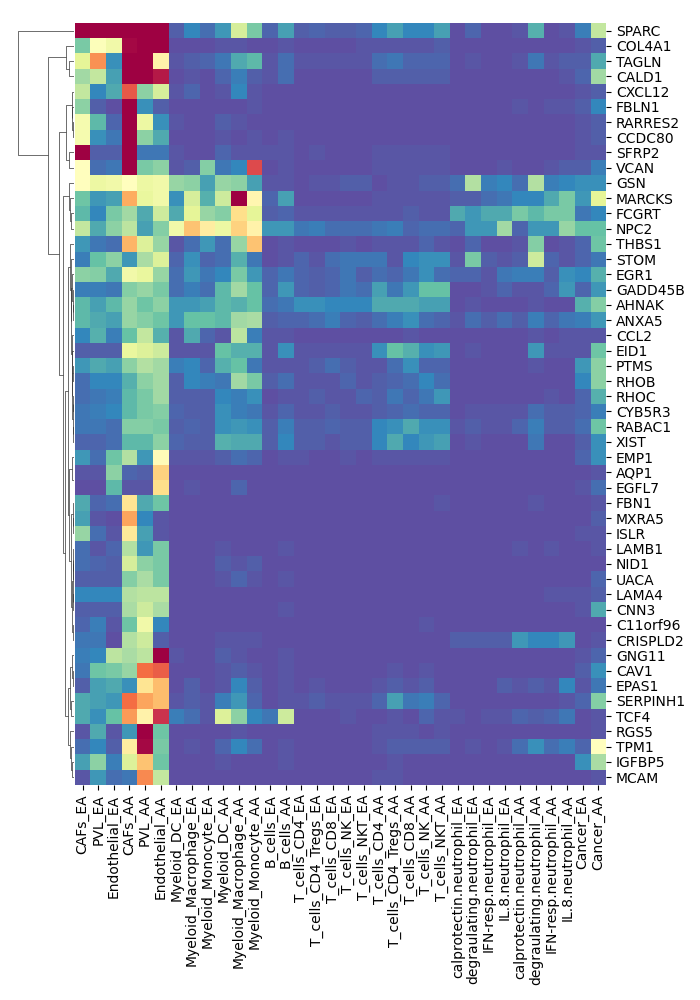
step3.do.pearson.2.bygene.custom
import math
import sys
import re
import os
import numpy as np
import matplotlib as mpl
import matplotlib.pyplot as plt
import scipy
import scipy.stats
from scipy.stats import zscore
import phenograph
from operator import itemgetter
import pandas as pd
import seaborn as sns
import pick_genes_by_total
def get_unique_clusters(n):
m = []
f = open(n)
for l in f:
l = l.rstrip("\n")
m.append(l)
f.close()
return m
#def read(n, order_n, gene_n):
def read(n, order_n, gene_file=None, genes_interest=None):
if gene_file is not None:
genes_interest = []
f = open(gene_file)
for l in f:
l = l.rstrip("\n")
genes_interest.append(l)
f.close()
f = open(n)
num_gene = 0
h = f.readline().rstrip("\n").split("\t")
for l in f:
l = l.rstrip("\n")
num_gene+=1
f.close()
f = open(n)
num_col = len(h) - 1
mat = np.zeros((num_gene, num_col), dtype="float32")
f.readline()
genes = []
gene_id = 0
for l in f:
l = l.rstrip("\n")
ll = l.split("\t")
t_gene = ll[0]
genes.append(t_gene)
mat[gene_id, :] = [float(v) for v in ll[1:]]
gene_id+=1
f.close()
header = h[1:]
header = np.array(header)
m_h = {}
for t_id, val in enumerate(header):
m_h[val] = t_id
f = open(order_n)
cond = []
new_order = []
for l in f:
l = l.rstrip("\n")
cond.append(l)
new_order.append(m_h[l])
f.close()
new_order = np.array(new_order)
mat = mat[:, new_order]
gmap = {}
for ix,v in enumerate(genes):
gmap[v] = ix
good_ids = np.array([gmap[g] for g in genes_interest])
mat = mat[good_ids, :]
genes = np.array(genes)
genes = genes[good_ids]
return mat, genes, header[new_order]
def is_not_filtered(g):
if g.startswith("RPL") or g.startswith("RPS") or g.startswith("HLA-"):
return False
else:
return True
if __name__=="__main__":
choice = sys.argv[2] #AA or EA
_, _, _, gene_sum = pick_genes_by_total.read("cell.type.expr.%s.%s.more.joined.txt" % (sys.argv[1], choice), choice)
#g,g_CAF,g_T,g_Myeloid,g_Neutrophil, g_T+g_Neutrophil
if choice=="EA":
gene_sum.sort(key=itemgetter(1), reverse=True)
genes_AA = set([g[0] for g in gene_sum[:35]])
gene_sum.sort(key=itemgetter(5), reverse=True) #important
gene_sum = [g for g in gene_sum if g[0] not in genes_AA]
else:
gene_sum.sort(key=itemgetter(5), reverse=True)
genes_EA = set([g[0] for g in gene_sum[:35]])
gene_sum.sort(key=itemgetter(1), reverse=True) #important
gene_sum = [g for g in gene_sum if g[0] not in genes_EA]
g_of_interest = [g[0] for g in gene_sum if is_not_filtered(g[0])]
g_of_interest = g_of_interest[:50]
#g_of_interest = [g[0] for g in gene_sum]
#g_of_interest = ["C1QB", "COL1A2", "DCN", "COL4A2", "THY1", "IGFBP7", "FCER1G", "IGKC", "FN1", "VCAN", "PCOLCE", "FSTL1", "ITM2B", "CTSB", "S100A4"]
#g_of_interest = ["S100A9", "PLSCR1", "CD6", "CAPG", "EFHD2", "RAC2", "CXCL9", "BIRC3", "CTSC", "GZMK", "CCL5", "IL2RG", "LY6E", "CD48", "CD2", "MX1", "IFITM1"]
mat, genes, conditions = read("cell.type.expr.%s.%s.more.joined.txt" % (sys.argv[1], choice), "order.%s.query.2.txt" % choice, genes_interest=g_of_interest, gene_file=None) #good
sys.stdout.write("\t".join(conditions) + "\n")
for ig,g in enumerate(genes):
sys.stdout.write(g + "\t" + "\t".join(["%.3f" % v for v in mat[ig,:]]) + "\n")
#plt.tick_params(axis='both', which='major', labelsize=10, labelbottom = False, bottom=False, top = False, labeltop=True)
nj_union = genes
nj_union_title = []
for n in nj_union:
#n_new = n.split(" ")[0]
n_new = n
nj_union_title.append(n_new)
nt = {}
for ind,ki in enumerate(conditions):
nx = []
for ind2, g in enumerate(genes):
#es = sum_values[protein_map[g], ind]
nx.append((ki, g, mat[ind2, ind], mat[ind2, ind]))
#ki_new = ki.split(" ")[0]
ki_new = ki
nt[ki_new] = pd.Series([tx[2] for tx in nx], index=nj_union_title)
cg=sns.clustermap(pd.DataFrame(nt), row_cluster=True, col_cluster=False, \
#7,10
figsize=(7, 10), method="ward", \
#vmax=3, vmin=0, #AA \
#vmax=2, vmin=0, #AA \
vmax=0.7, vmin=0, #EA, good \
#vmax=1.0, vmin=0, #EA \
#col_ratios={"dendrogram":0.05}, \
#row_ratios={"dendrogram":0.05}, \
dendrogram_ratio=(0.1, 0.01), \
cbar_pos=None,\
#cmap="rainbow", \
cmap="Spectral_r", \
#cmap="Reds", \
yticklabels=True, xticklabels=True)
#plt.show()
#sys.exit(0)
#cg.fig.savefig("heatmap_%s.png" % (t_key))
#cg.ax_heatmap.tick_params(labeltop=True, labelbottom=False, labelleft=True, labelright=False, \
#top=True, bottom=False, left=True, right=False)
plt.setp(cg.ax_heatmap.yaxis.get_majorticklabels(), rotation=0)
#save_file = "heatmap_%s.png" % (sys.argv[1])
#save_file = "heatmap_AAcoef_%s.png" % sys.argv[1]
save_file = "heatmap_%scoef_%s.png" % (choice, sys.argv[1])
#save_file = "heatmap_AAcoef.png"
#save_file = "heatmap_EAcoef_rearr.png"
#save_file = "heatmap_AAcoef_rearr.png"
cg.fig.savefig("%s" % save_file)
sys.exit(0)
'''
protein_map = {}
for ind,v in enumerate(proteins):
protein_map[v] = ind
by_category = read_case_map("pngs/sheet")
print(by_category)
'''
#print(by_case)
#sys.exit(0)
#group1 = ["Case10_ROI002", "Case10_ROI003", "Case10_ROI004"]
#group1 = ["Case10_ROI005", "Case10_ROI006", "Case10_ROI007"]
#group1 = ["Case10_ROI001", "Case10_ROI008"]
#group1 = ["Case11_ROI001", "Case11_ROI007"]
#group1 = ["Case11_ROI003", "Case11_ROI004", "Case11_ROI005", "Case11_ROI006"]
#group1 = ["Case11_ROI002"]
#group1 = ["Case1_ROI001", "Case1_ROI003", "Case1_ROI004"]
#group1 = ["Case1_ROI002", "Case1_ROI005", "Case1_ROI006"]
for t_category in by_category:
print(t_category)
group1 = by_category[t_category]
sum_values = np.zeros((len(proteins), len(proteins)), dtype="float32")
for t_key in group1:
t_case = by_case[t_key]
values = np.zeros((len(proteins), len(proteins)), dtype="float32")
for i in range(len(proteins)):
for j in range(len(proteins)):
values[i,j] = scipy.stats.pearsonr(t_case[:,i], t_case[:,j])[0]
sum_values[i,j] += 1/len(group1) * scipy.stats.pearsonr(t_case[:,i],t_case[:,j])[0]
nj_union = proteins
nj_union_title = []
for n in nj_union:
n_new = n.split("_")[2]
nj_union_title.append(n_new)
nt = {}
for ind,ki in enumerate(proteins):
nx = []
for g in nj_union:
es = sum_values[protein_map[g], ind]
nx.append((ki, g, np.mean(es), np.std(es)))
#cluster_expr[:, ki-1] = np.array([n[2] for n in nx])
ki_new = ki.split("_")[2]
nt[ki_new] = pd.Series([tx[2] for tx in nx], index=nj_union_title)
cg=sns.clustermap(pd.DataFrame(nt), row_cluster=False, col_cluster=False, \
figsize=(5, 5), method="average", \
vmax=1.0, vmin=-1.0, \
#col_ratios={"dendrogram":0.05}, \
#row_ratios={"dendrogram":0.05}, \
dendrogram_ratio=(0.1, 0.01), \
#cbar_pos=None,\
cmap="Spectral", yticklabels=True, xticklabels=True)
plt.setp(cg.ax_heatmap.yaxis.get_majorticklabels(), rotation=0)
#plt.show()
#cg.fig.savefig("heatmap_%s.png" % (t_key))
save_file = "heatmap_%s_%s.png" % (t_category[0], t_category[1])
cg.fig.savefig("%s" % save_file)
Running Command
python3 step3.do.pearson.2.bygene.custom.py M1 AAResults